






Atomic spectrum #2: Custom broadening function¶
The lbfunc parameter of SpectrumFactory allows us to specify a custom function to use for the Lorentzian broadening of a spectrum. This is especially useful for handling pressure broadening of atomic lines for which multiple theories exist.
By default, RADIS calculates the total Lorentzian broadening of atomic lines as the sum of the Van der Waals broadening, Stark broadening, and radiation broadening as returned by gamma_vald3() from the broadening parameters for each of these types provided in the databank. When the databank doesn’t provide these parameters (as is the case with NIST), lbfunc becomes a required argument as there is no default to cater for this case.
The line shift is also calculated by default as 2/3 times the Van der Waals broadening, and lbfunc allows you to specify an alternative line shift as the 2nd return argument if you wish.
lbfunc can be changed on the fly by changing the sf.params.lbfunc attribute of the SpectrumFactory instance, the result of which is reflected next time the library calculates its Lorentzian broadening.
To make use of it, familiarise yourself with the column names that RADIS assigns to the relevant quantities you intend to use in lbfunc.
from radis import SpectrumFactory
def lbfunc1(**kwargs):
return 0.1 * (296 / kwargs["Tgas"]) ** 0.7, None
mole_fraction = 0.01
sf = SpectrumFactory(
12850,
12870,
species="O_I",
pressure=1.01325, # = 1 atm
diluent={"H": 1 - mole_fraction - 1e-3, "e-": 1e-3}, # so it all adds up to 1
mole_fraction=mole_fraction,
path_length=15,
lbfunc=lbfunc1,
pfsource="barklem",
)
sf.fetch_databank("kurucz")
s1 = sf.eq_spectrum(4000)
/home/docs/checkouts/readthedocs.org/user_builds/radis/checkouts/latest/radis/misc/warning.py:443: UserWarning: The required columns for Kurucz don't match those of existing moleculear databases, so all columns are being loaded
warnings.warn(WarningType(message))
Attempting to download http://kurucz.harvard.edu/atoms/0800/gf0800.all
- Downloading gf0800.all (1/1)
Downloading: 0%| | 0.00/2.07M [00:00<?, ?B/s]
Downloading: 5%|▍ | 96.0k/2.07M [00:00<00:02, 879kB/s]
Downloading: 18%|█▊ | 392k/2.07M [00:00<00:00, 1.94MB/s]
Downloading: 54%|█████▎ | 1.11M/2.07M [00:00<00:00, 4.15MB/s]
Downloading: 100%|██████████| 2.07M/2.07M [00:00<00:00, 5.48MB/s]
Successfully downloaded http://kurucz.harvard.edu/atoms/0800/gf0800.all
Added Kurucz-O_I database in /home/docs/radis.json
0.68s - Loaded database
/home/docs/checkouts/readthedocs.org/user_builds/radis/checkouts/latest/radis/misc/warning.py:443: UserWarning: wavenumber shift not given in database: assumed 0 shift
warnings.warn(WarningType(message))
Calculating Equilibrium Spectrum
Physical Conditions
----------------------------------------
Tgas 4000 K
isotope 0
medium air
mole_fraction 0.01
path_length 15 cm
pressure 1.01325 bar
self_absorption True
species O_I
state X
wavenum_max 12870.0000 cm-1
wavenum_min 12850.0000 cm-1
Computation Parameters
----------------------------------------
Tref 296 K
add_at_used numpy
broadening_method voigt_poly
cutoff 0 cm-1/(#.cm-2)
dbformat kurucz
dbpath /home/docs/.radisdb/kurucz/O_I-gf0800.h5
folding_thresh 1e-06
include_neighbouring_lines True
isatom True
isneutral True
lbfunc <function lbfunc1 at 0x71bd6c1a59b0>
memory_mapping_engine auto
neighbour_lines 0 cm-1
optimization simple
parsum_mode full summation
pfsource barklem
potential_lowering None
pseudo_continuum_threshold 0
sparse_ldm True
truncation 50 cm-1
waveunit cm-1
wstep 0.01 cm-1
zero_padding 2002
----------------------------------------
0.01s - Spectrum calculated
Now compare the result with that of the default handling of broadening in RADIS by removing the lbfunc parameter, recalculating the spectrum without it, and plotting the diff between the results:
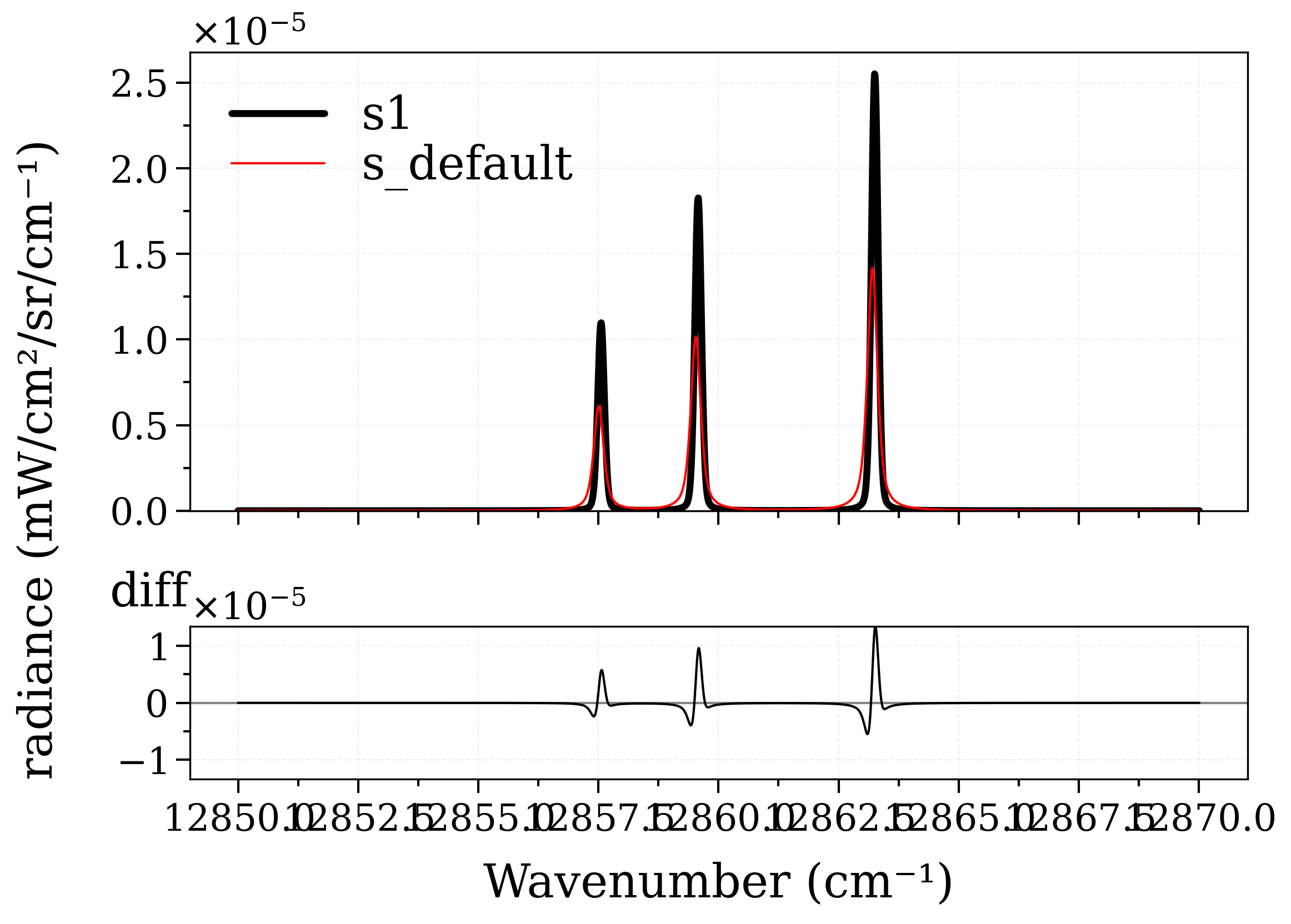
Calculating Equilibrium Spectrum
Physical Conditions
----------------------------------------
Tgas 4000 K
isotope 0
medium air
mole_fraction 0.01
path_length 15 cm
pressure 1.01325 bar
self_absorption True
species O_I
state X
wavenum_max 12870.0000 cm-1
wavenum_min 12850.0000 cm-1
Computation Parameters
----------------------------------------
Tref 296 K
add_at_used numpy
broadening_method voigt_poly
cutoff 0 cm-1/(#.cm-2)
dbformat kurucz
dbpath /home/docs/.radisdb/kurucz/O_I-gf0800.h5
folding_thresh 1e-06
include_neighbouring_lines True
isatom True
isneutral True
lbfunc None
memory_mapping_engine auto
neighbour_lines 0 cm-1
optimization simple
parsum_mode full summation
pfsource barklem
potential_lowering None
pseudo_continuum_threshold 0
sparse_ldm True
truncation 50 cm-1
waveunit cm-1
wstep 0.01 cm-1
zero_padding 2002
----------------------------------------
0.02s - Spectrum calculated
(<Figure size 640x480 with 2 Axes>, [<Axes: >, <Axes: xlabel='Wavenumber (cm⁻¹)'>])
Here’s another example adding self broadening, calculated using eq(16) of [Minesi-et-al-2020], to the 3 broadening types already handled by RADIS, and keeping the default handling of the line shift:
from radis.lbl.broadening import gamma_vald3
from radis.phys.constants import k_b_CGS
def lbfunc2(
df, pressure_atm, mole_fraction, Tgas, diluent, isneutral, **kwargs
): # assign variable names to the quantities we use and put the rest in kwargs which can be ignored
beta = 1e-4 # example
n_emitter = pressure_atm * 1.01325 * 1e6 * mole_fraction / (k_b_CGS * Tgas)
gamma_self = ((1e7 / df["wav"]) ** 2) * beta * n_emitter / 2.7e19
print(gamma_self)
# copied from default code in RADIS:
gammma_rad, gamma_stark, gamma_vdw = gamma_vald3(
Tgas,
pressure_atm * 1.01325,
df["wav"],
df["El"],
df["ionE"],
df["gamRad"],
df["gamSta"],
df["gamvdW"],
diluent,
isneutral,
)
print(gammma_rad, gamma_stark, gamma_vdw)
shift = (
(1.0 / 3.0) * 2 * gamma_vdw
) # Konjević et al. 2012 §4.1.3.2, neglect stark shift by default
wl = gammma_rad + gamma_stark + gamma_vdw + gamma_self
return wl, shift
sf.params.lbfunc = lbfunc2
s2 = sf.eq_spectrum(4000)
plot_diff(s2, s_default, label1="s2", label2="s_default")

0 0.041105
1 0.041092
2 0.041084
3 0.041068
Name: wav, dtype: float64
[0.00010327 0.00010327 0.00094182 0.00010327] 0 0.006780
1 0.006780
2 0.007099
3 0.006780
Name: gamSta, dtype: float64 [0.07269791 0.07269791 0.07269791 0.07269791]
Calculating Equilibrium Spectrum
Physical Conditions
----------------------------------------
Tgas 4000 K
isotope 0
medium air
mole_fraction 0.01
path_length 15 cm
pressure 1.01325 bar
self_absorption True
species O_I
state X
wavenum_max 12870.0000 cm-1
wavenum_min 12850.0000 cm-1
Computation Parameters
----------------------------------------
Tref 296 K
add_at_used numpy
broadening_method voigt_poly
cutoff 0 cm-1/(#.cm-2)
dbformat kurucz
dbpath /home/docs/.radisdb/kurucz/O_I-gf0800.h5
folding_thresh 1e-06
include_neighbouring_lines True
isatom True
isneutral True
lbfunc <function lbfunc2 at 0x71bd6f480ca0>
memory_mapping_engine auto
neighbour_lines 0 cm-1
optimization simple
parsum_mode full summation
pfsource barklem
potential_lowering None
pseudo_continuum_threshold 0
sparse_ldm True
truncation 50 cm-1
waveunit cm-1
wstep 0.01 cm-1
zero_padding 2002
----------------------------------------
0.02s - Spectrum calculated
(<Figure size 640x480 with 2 Axes>, [<Axes: >, <Axes: xlabel='Wavenumber (cm⁻¹)'>])
We can even modify broadening parameters of individual lines:
def lbfunc3(df, Tgas, pressure_atm, diluent, isneutral, **kwargs):
# only for Pandas dataframes:
df.loc[df["orig_wavelen"] == 777.5388, "gamvdW"] = (
-7
) # should also result in a different shift
df.loc[df["orig_wavelen"] == 777.1944, "gamSta"] = -5
# copied from default code in RADIS:
gammma_rad, gamma_stark, gamma_vdw = gamma_vald3(
Tgas,
pressure_atm * 1.01325,
df["wav"],
df["El"],
df["ionE"],
df["gamRad"],
df["gamSta"],
df["gamvdW"],
diluent,
isneutral,
)
shift = (1.0 / 3.0) * 2 * gamma_vdw # Konjević et al. 2012 §4.1.3.2
wl = gammma_rad + gamma_stark + gamma_vdw
return wl, shift
sf.params.lbfunc = lbfunc3
s3 = sf.eq_spectrum(4000)
plot_diff(s3, s_default, label1="s3", label2="s_default")
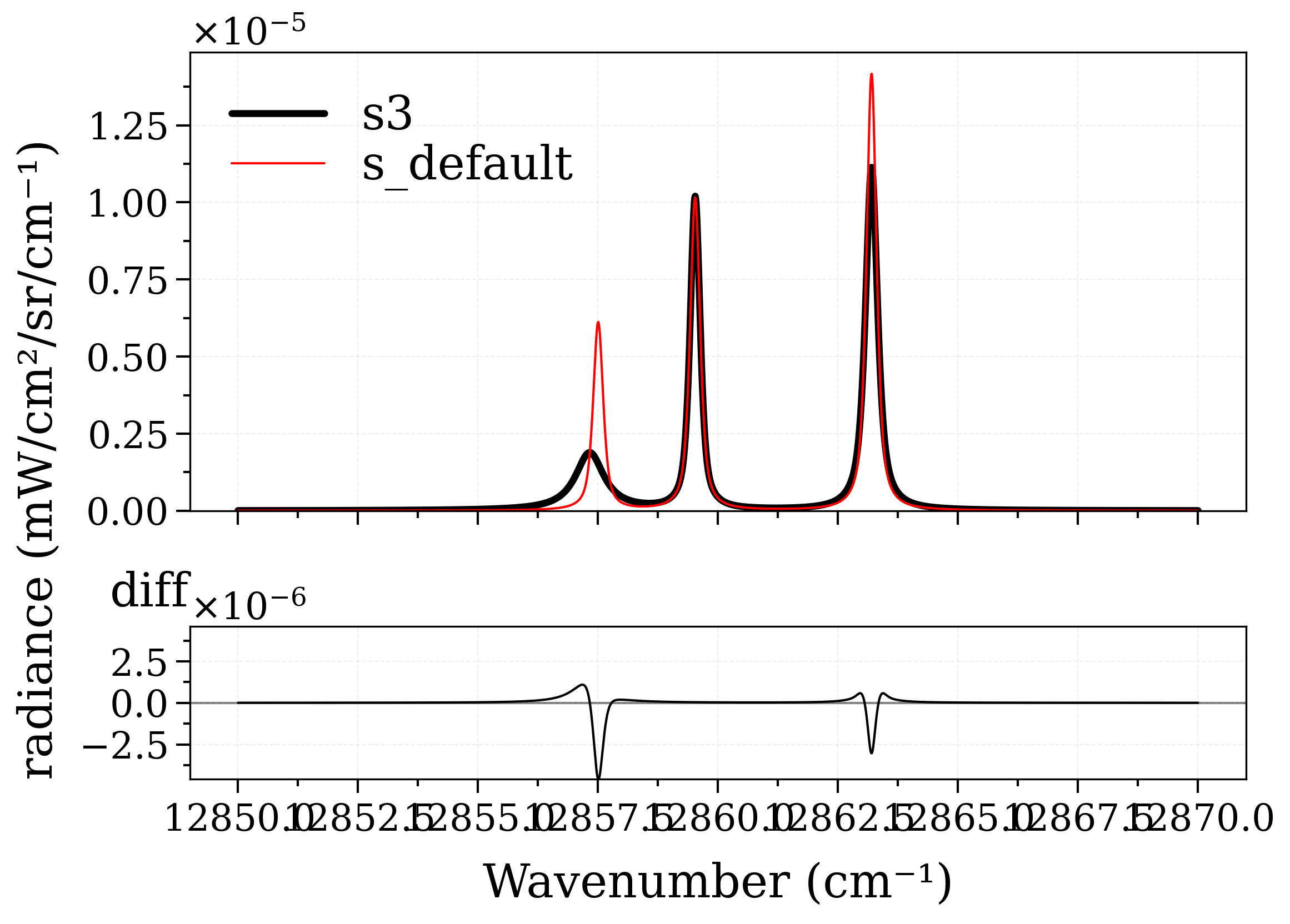
Calculating Equilibrium Spectrum
Physical Conditions
----------------------------------------
Tgas 4000 K
isotope 0
medium air
mole_fraction 0.01
path_length 15 cm
pressure 1.01325 bar
self_absorption True
species O_I
state X
wavenum_max 12870.0000 cm-1
wavenum_min 12850.0000 cm-1
Computation Parameters
----------------------------------------
Tref 296 K
add_at_used numpy
broadening_method voigt_poly
cutoff 0 cm-1/(#.cm-2)
dbformat kurucz
dbpath /home/docs/.radisdb/kurucz/O_I-gf0800.h5
folding_thresh 1e-06
include_neighbouring_lines True
isatom True
isneutral True
lbfunc <function lbfunc3 at 0x71bd66a4ba00>
memory_mapping_engine auto
neighbour_lines 0 cm-1
optimization simple
parsum_mode full summation
pfsource barklem
potential_lowering None
pseudo_continuum_threshold 0
sparse_ldm True
truncation 50 cm-1
waveunit cm-1
wstep 0.01 cm-1
zero_padding 2002
----------------------------------------
0.03s - Spectrum calculated
(<Figure size 640x480 with 2 Axes>, [<Axes: >, <Axes: xlabel='Wavenumber (cm⁻¹)'>])
References¶
Total running time of the script: (0 minutes 2.260 seconds)